about.
This is a collection of applications for genomics data processing, primarily high-throughput next-generation sequencing. There is a focus on processing data in Wiggle format, since many other tools are available for SAM/BAM (1,2), Bed (1,2), Fastq, etc. Wiggle/BigWig formats provide a compact way to store numerical data resulting from ChIP-seq, MNase-seq, FAIRE-seq, and DNase-seq experiments. This toolkit provides applications for adding, subtracting, dividing, multiplying, log-transforming, averaging, Z-scoring, and smoothing Wig files. There are also tools for performing analysis of MNase-seq (nucleosome mapping) data, creating heatmaps, and averaging the values from aligned loci.
Tools may be run from the terminal or from Galaxy.
All tools are designed to process data in chunks so that memory requirements never exceed ~1GB, regardless of genome size. Tools are intended to be modular, so that multiple tools can easily be strung together into ad hoc pipelines or workflows in Galaxy. For example, a common pipeline for our ChIP-seq experiments is: 1) map reads with bowtie, 2) calculate coverage of sequencing reads, 3) normalize by subtracting input, 4) Z-score the normalized coverage, 5) correlate replicates, 6) average multiple replicates, and 7) make a heatmap of the final result.
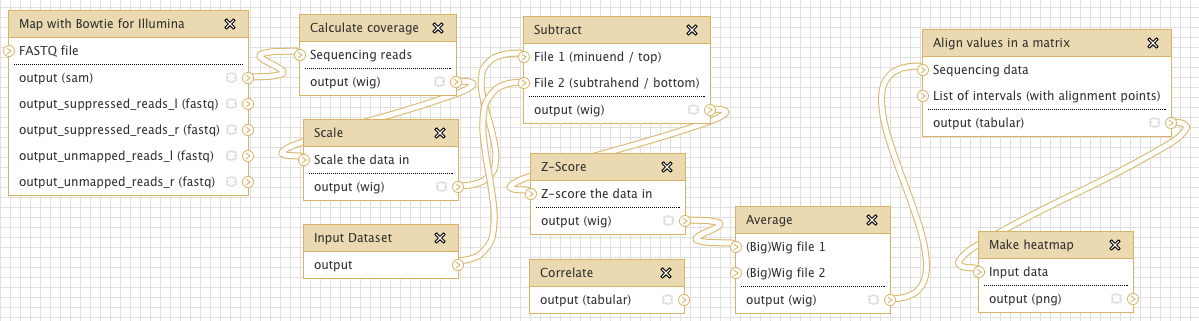
tools.
For an up-to-date list of available tools, search for java-genomics-toolkit in the Galaxy Tool Shed.
converters.
- IntervalToWig: convert valued Bed/BedGraph/GFF interval data (i.e. from microarrays) to Wiggle format
- IntervalToBed: convert valued BigBed/BedGraph/GFF/VCF/SAM/BAM reads or intervals to Bed format
- RomanNumeralize: convert chromosome names to Roman Numerals (i.e. chr12 to chrXII)
- GeneTrackToWig: convert GeneTrack formatted data to Wiggle format
- GeneTrackToBedGraph: convert GeneTrack formatted data to BedGraph format
- InterpolateWig: interpolate missing values in a (Big)Wig file (useful for making continuous tracks from microarray data)
- FastqIlluminaToSanger: quickly convert Illumina-encoded Phred scores in a FASTQ file to Sanger format
dna.
- DNAPropertyCalculator: calculate sequence-specific DNA properties, such as GC content, Roll, Twist, etc.
- FindNMers: find matches to a short sequence in a reference genome
ngs.
- Autocorrelation: compute the autocorrelation function of Wiggle data intervals
- BaseAlignCounts: generate occupancy tracks in Wiggle format from mapped reads in SAM/BAM/Bed format
- FindAbsoluteMaxima: finds absolute maxima of Wiggle data in intervals
- FindOutlierRegions: find potential CNVs or deletions from sequencing coverage data
- IntervalLengthDistribution: computes a histogram of interval/read lengths
- IntervalStats: computes mean/min/max values of Wiggle data over a set of intervals
- PowerSpectrum: computes the power spectrum of Wiggle data over a set of intervals
- RollingReadLength: generates a Wiggle track with the mean read length over each base pair
- ReadLengthDistributionMatrix: examine paired-end read length distribution across an interval of the genome
- WaveletTransform: compute a Wavelet scaleogram across an interval of the genome
- ExtractDataFromRegion: get data for a region from (Big)Wig file(s)
nucleosomes.
- FindBoundaryNucleosomes: finds the 5' and 3' nucleosomes for a set of intervals
- GreedyCaller: calls stereotypic nucleosome positions with a greedy algorithm
- MapDyads: maps read centers / approximated nucleosome positions into a Wiggle track
- Phasogram: computes the distribution of read phases (distance between reads)
- PercusDecomposition: compute a Percus energy decomposition from nucleosome data
- PredictFAIRESignal: attempt to model FAIRE experiment data from nucleosome occupancy
- DynaPro: solve single-particle statistical mechanics equilibria using a dynamic programming algorithm
visualization.
- IntervalAverager: computes the average profile for a set of aligned intervals
- KMeans: k-means clustering of a matrix of data
- MatrixAligner: aligns intervals of Wiggle data into a matrix for visualization with matrix2png
- StripMatrix: strips the headers off of a matrix2png-format matrix for import into Matlab/R
wigmath.
- Add: add multiple (Big)Wig files
- Subtract: subtract two (Big)Wig files
- Multiply: multiply (Big)Wig files together
- Divide: divide two (Big)Wig files
- Average: average multiple (Big)Wig files
- ZScore: a (Big)Wig file
- LogTransform: log-transform a (Big)Wig file
- MovingAverageSmooth: smooth a (Big)Wig file with a box (moving average) kernel
- GaussianSmooth: smooth a (Big)Wig file with a Gaussian kernel
- Scale: scale a (Big)Wig file by a normalization constant
- MeanShift: a (Big)Wig file to have a specified mean
- Correlate: multiple (Big)Wig files (Pearson/Spearman)
- ValueDistribution: compute a histogram of values in a (Big)Wig file
- Summary: generate summary descriptive statistics about a (Big)Wig file
- Downsample: a (Big)Wig file into windows
usage.
galaxy.
One-click installation is available for your local Galaxy instance through the Galaxy Tool Shed.
If you run a production Galaxy server, configuration files are provided for loading the applications into Galaxy manually. Unzip or check out the java-genomics-toolkit distribution into Galaxy's "tools" folder, and add the supplied tool_conf entries to your tool_conf.xml file.
shell.
Tools can also be run on the terminal, and helper scripts are provided for convenience. For more information and usage examples, see the GitHub page.
requirements.
java-genomics-toolkit requires Java 7, available at oracle.com.
download.
The recommended way to obtain the toolkit is to check out the source code from GitHub and build it using the provided Ant build script (simply call "ant").
In addition, precompiled, ready-to-use packages that include the JRE v7 are available for Linux platforms in x32 and x64 flavors. If you want to try out the toolkit, this may be the quickest option.
to.do.
- Parallelize: explore potential performance benefits of parallelizing the computation across multiple processes / cluster nodes
java-genomics-io
Those wishing to write their own scripts may be interested in java-genomics-io, the library upon which these applications are built. This library supports iterating or querying for data from Bed, BedGraph, GeneTrack, GFF, SAM, BAM, Wiggle, BigWig, and BigBed files with a consistent interface. ASCII files are indexed with Tabix as needed to perform queries efficiently. Writers are also available for writing Bed, BedGraph, GFF, SAM, BAM, and Wig files.
licensing.info.
java-genomics-toolkit is distributed under the GNU General Public License v3. See the included license.txt for more details.
java-genomics-toolkit was created by Timothy Palpant for work in the Lieb laboratory at UNC Chapel Hill.
java-genomics-toolkit utilizes multiple external libraries including:
- java-genomics-io to parse genomic data files
- BigWig/BigBed Reader (by the Broad Institute) to read BigWig/BigBed files
- Picard / SAM-JDK for reading SAM/BAM files
- JCommander for processing command-line arguments
- JTransforms for FFT
- log4j for logging
- commons-lang for String and Array manipulation
- commons-math for mathematics
contact.me.
Please contact me at tim [at] palpant.us with bugs, questions, comments, or suggestions.